О проекте
Лаборатория функциональной геномики уже несколько лет успешно занимается разработкой подходов и проведением функциональных исследований вариантов нуклеотидной последовательности.
Мы помогли разрешить с клинической точки зрения десятки случаев. Проведенные нами исследования позволили доказать патогенность вариантов неопределённого клинического значения как в генах, ранее описанных для наследственных заболеваний, так и в генах, прежде не ассоциированных с наследственной патологией.
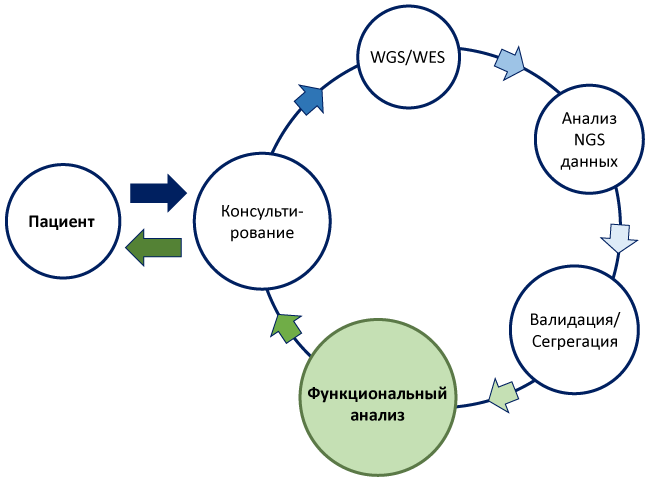
Функциональный анализ вариантов нуклеотидной последовательности
Центральной проблемой при проведении ДНК-диагностики наследственных заболеваний является определение патогенности выявленных вариантов нуклеотидной последовательности. Сегодня оно осуществляется согласно российскому Руководству по интерпретации данных, полученных методами массового параллельного секвенирования (MPS).
Однако при первичном анализе данных MPS, у биоинформатика часто не хватает информации для признания того или иного варианта нуклеотидной последовательности патогенным. В таких случаях для установления диагноза пациенту врач-генетик может рекомендовать проведение функционального анализа выявленных вариантов. Данные исследования должны быть валидируемыми, воспроизводимыми и проведенными в соответствии со строгими правилами постановки научного эксперимента.
Для оценки патогенности/доброкачественности вариантов нуклеотидной последовательности в лаборатория функциональной геномики был создан калькулятор патогенности вариантов нуклеотидной последовательности
В лаборатории функциональной геномики «Медико-генетического научного центра» уже несколько лет занимаются разработкой подходов и проведением функциональных исследований вариантов нуклеотидной последовательности. Мы помогли разрешить с клинической точки зрения десятки кейсов. Проведенные нами исследования позволили доказать патогенность вариантов неопределённого клинического значения как в генах, ранее описанных для наследственных заболеваний, так и в генах, прежде не ассоциированных с наследственной патологией.
На данном сайте вы сможете ознакомится с примерами функциональных исследований, выполненных в нашей лаборатории, и оставить свою заявку для рассмотрения возможности проведения функционального анализа обнаруженного вами варианта нуклеотидной последовательности.
Важность проведения функционального анализа
Существует много подходов к оценке патогенности ранее не описанных вариантов нуклеотидной последовательности такие как: анализ сегрегации в семье, использование популяционных данных, биоинформатические подходы и другие. Однако очень часто этого бывает недостаточно и вариант так и остается «вариантом неясного значения».
В таких случаях единственный способ достоверно убедиться в патогенности варианта – провести его функциональный анализ в системе in-vitro или in-vivo.
Его проведение не только дает уверенность в точной причине заболевания, что крайне необходимо для корректного медико-генетического консультирования семей и установления риска повторного рождения больного ребенка, что особенно актуально для генетически гетерогенных состояний, но и позволяет установить патогенетические механизмы, лежащие в основе заболевания, что может быть использовано в перспективе для подбора таргетной терапии пациентов с наследственной патологией.
Согласно отечественным и зарубежным руководствам крайне не рекомендуется проведение дородовой диагностики на «варианты неясного значения». Если отсутствует уверенность в патогенности варианта, то в результате даже после проведения дородовой диагностики существует риск рождения больного ребенка. В таких случаях лишь функциональный анализ может внести ясность, что является первостепенной задачей для семей, которые планируют продолжить деторождение.
Примеры работ
Функциональный анализ вариантов в гене COL6A3 приводящих к спектру миопатий от врожденной мышечной дистрофии Ульриха до миопатии Бетлема
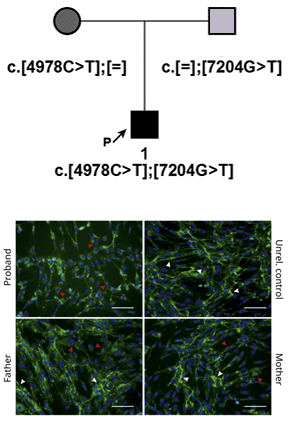
Врачами научно-консультативного отделения (НКО) МГНЦ» Дадали Еленой Леонидовной и Шарковой Инной Валентиновной в нашу лабораторию для проведения функционального анализа был направлен пациент (8 лет) с пограничным состоянием между тяжелой наследственной мышечной дистрофией Ульриха и более мягкой миопатией Бетлема. Ранее при полноэкзомном секвенировании у пробанда были выявлены неописанные изменения в нуклеотидной последовательности гена COL6A3: отец и мать являлись гетерозиготными носителями вариантов c.7204G>T (p.Glu2402Ter) и c.4978C>T (p.Arg1660Cys) соответственно, в то время как сын имел компаунд-гетерозиготное состояние данных вариантов. Целью нашей работы было определить патогенность выявленных вариантов нуклеотидной последовательности. Для этого всем членам семьи была проведена биопсии кожи, и получены первичные культуры фибробластов. На этих культурах был проведен анализ экспрессии гена COL6A3. В фибробластах пробанда была обнаружена экспрессия только материнского аллеля с заменой c.4978C>T. Экспрессия гена с отцовского аллеля c.7204G>T отсутствовала как у пробанда, так и у отца, что свидетельствовало в пользу нонсенс-опосредованной деградации мРНК (NMD).
Далее на первичных культурах фибробластов семьи было проведено иммунофлуоресцентное окрашивание белка COL6A3. Паттерн распределения COL6A3 в культуре фибробластов пробанда выявил внутриклеточную локализацию иммунореактивного материала, а также общее снижение уровня белка. Культуры фибробластов родителей демонстрировали присутствие как внутриклеточной (незрелые предшественники проколлагена), так и внеклеточной (фибриллярные) фракции COL6A3. Стоит отметить, что внеклеточный коллагеновый матрикс в культуре фибробластов отца был более выражен по сравнению с культурой фибробластов матери, что может говорить о легком доминантно-негативном эффекте миссенс замены p.Arg1660Cys. Тем не менее, ни в одном из случаев коллаген не был организован в тонкую фибриллярную сеть, как в случае контрольной культуры фибробластов. Детальное фенотипирование клинически здоровых родителей выявило субклинические признаки дисплазии соединительной ткани, более выраженные у матери. Таким образом, экспериментальные данные находятся в соответствии с клиническими проявлениями и подтверждают патогенное значение ранее не описанных вариантов c.7204G>T и c.4978C>T в гене COL6A3.
Поиск патогенного варианта в гене PALB2 методом экспрессионного анализа
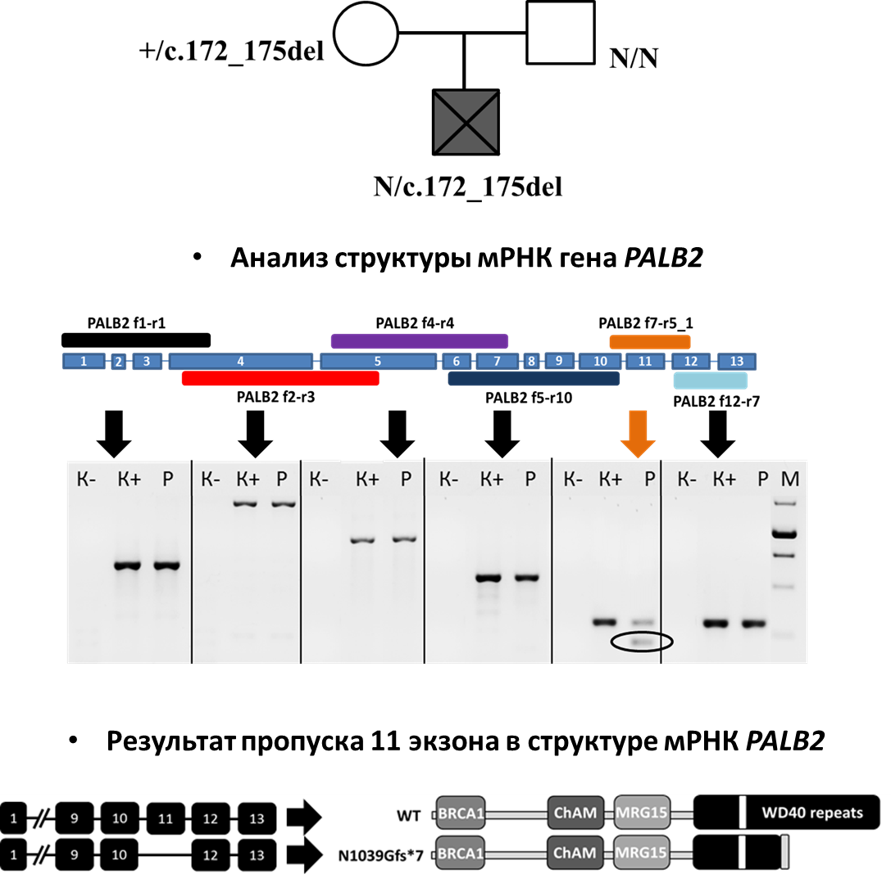
В лабораторию обратилась врач МГНЦ Бессонова Людмила Александровна с просьбой выявить вторую мутацию в гене PALB2 при анемии Фанкони у ребёнка в семье, проходившей консультацию и разработку ПГД в лаборатории «Генетико». При секвенировании экзома ребёнка был обнаружен только один описанный раннее патогенный вариант c.172_175del, унаследованный от матери. Отцу было также проведено секвенирование экзома в лаборатории «Геномед», однако значимых вариантов не было обнаружено. MLPA анализ ДНК отца также не выявил структурных нарушений в гене PALB2.
Биоинформатиком Ф.А.Коноваловым была выдвинута гипотеза, что второй вариант может находиться в интроне и влиять на сплайсинг или экспрессию гена PALB2. Для установления аномальной структуры мРНК гена PALB2 её последовательность была разделена на 6 перекрывающихся участков, структура каждого из которых была проанализирована методом ПЦР с обратной транскрипцией. Для анализа использовали РНК, выделенную из крови отца пробанда. В результате, был обнаружен аномальный транскрипт с пропуском 11 экзона. Для дальнейшего поиска интронного варианта в гене PALB2 было проведено секвенирование по Сенгеру геномной ДНК отца в районе 11 экзона. Таким способом была выявлена гетерозиготная делеция в 10 интроне c.3114-16_3114-11del. Её влияние на структуру мРНК гена PALB2 было подтверждено с использованием системы экспрессии минигена.
Пропуск 11 экзона приводит к сдвигу рамки считывания и как следствие к образованию белка с укороченным WD40 доменом. По данным литературы, белок с подобной структурой не является функциональным.
Функциональный анализ варианта c.5655+5G>C в гене MYH7
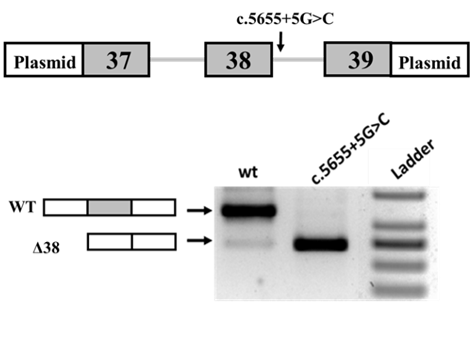
В лабораторию за анализом варианта c.5655+5G>C в гене MYH7 обратилась заведующая лабораторией медицинской генетики РНЦХ Заклязьминская Елена Валерьевна. Данный вариант был выявлен при полногеномном секвенировании ДНК пациента мужского пола (1,5 лет) с врожденной слабостью осевой мускулатуры, синдромом «свисающей головы» и дилатационной кардиомиопатией. В лаборатории медицинской генетики РНЦХ было установлено, что данный вариант является de novo вариантом. Кроме того, ОТ-ПЦР анализ РНК, выделенной из крови пациента, показал наличие двух изоформ мРНК MYH7: нормальной и с пропуском экзона 38.
Для подтверждения того, что вариант c.5655+5G>C влияет на сплайсинг пре-мРНК, нами была использована система экспрессии минигена MYH7 в модельной клеточной линии. В ходе работы было установлено, что исследуемый вариант приводит к in-frame пропуску экзона 38. На уровне белка данные изменения приводят к делеции 32 аминокислот в C-терминальном домене MYH7 (p.1854_1885del). По литературным данным такое изменение белка приводит к развитию заболевания. Таким образом, нами была подтверждена патогенность ранее не описанного варианта c.5655+5G>C в гене MYH7.
Результаты данной работы рассматриваются в виде публикации в журнале Gene
Функциональный анализ вариантов, влияющих на сплайсинг, в гене PAX6
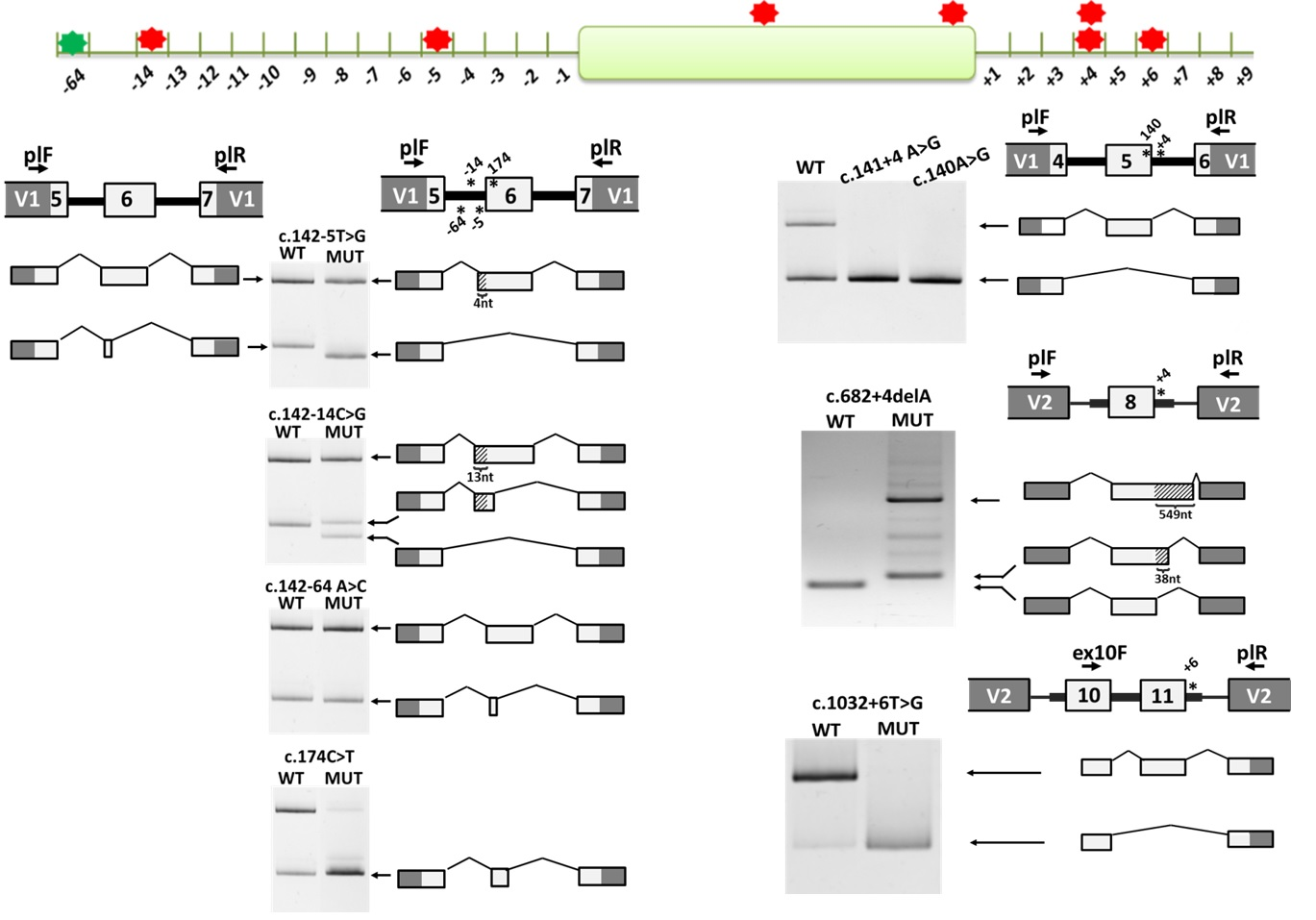
В нашу лабораторию обратилась научный сотрудник лаборатории генетической эпидемиологии МГНЦ Васильева Татьяна Алексеевна. Ранее в лаборатории генетической эпидемиологии было проведено исследование 177 пациентов с диагнозом врожденная аниридия из 144 семей Российской Федерации методами MLPA и прямого секвенирования по Сенгеру гена PAX6. В результате было выявлено 96 вариантов в гене PAX6, 20 из которых предположительно влияют на сплайсинг. 10 из этих вариантов находились в положениях ±1/2, 8 – в других областях интрона. Кроме того, были выявлены одна миссенс и одна синонимичная замены, которые предположительно влияют на сплайсинг. Нашей задачей стало исследование предположительного влияния выявленных вариантов на сплайсинг пре-мРНК PAX6.
Нами было проведено исследование 6 интронных вариантов в гене PAX6, находящихся вне позиций ±1/2, а также одной синонимичной и одной миссенс замены с использованием системы экспрессии минигена в модельной клеточной линии. В ходе работы нами было установлено, что 7 вариантов оказывают влияние на сплайсинг пре-мРНК PAX6, в том числе миссенс (c.140A>G) и синонимичный (c.174C>T) варианты, тогда как один из интронных вариантов (c.142-64A>C) не показывает эффекта на сплайсинг. Все выявленные изменения структуры мРНК приводят к образованию преждевременного стоп-кодона и запуску механизма нонсенс-опосредованной деградации (NMD) мРНК, что в результате и приводит к гаплонедостаточности функции гена PAX6. Таким образом, нами было выявлено, что доля мутаций сплайсинга в гене PAX6 при врожденной аниридии значительно выше, чем было принято считать ранее. В результате данной работы, удалось установить точный молекулярно-генетический диагноз 6 пациентам.
Функциональный анализ варианта с.193+5G>A в гене C19orf12
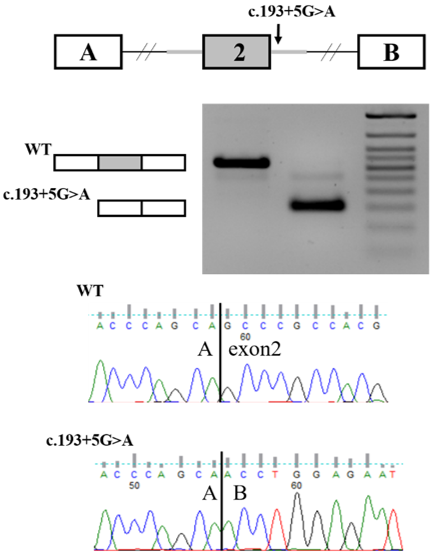
В лабораторию обратилась врач МГНЦ Шаркова Инна Валентиновна с просьбой оценить патогенность выявленного варианта нуклеотидной последовательности с.193+5G>A в гене C19orf12 у больной с клинической картиной спастического парапареза, выраженной постуральной неустойчивости, прогрессирующим снижением зрения, нарушения поведения и МРТ признаками накопления железа в подкорковых структурах. Данный вариант был выявлен в ходе секвенирования панели генов "Нейродегенеративные заболевания" в лаборатории «ГЕНОМЕД». Так же в гене C19orf12 была выявлена ранее описанная патогенная делеция. Частота варианта с.193+5G>A согласно контрольным выборкам крайне низкая, программы предсказания оценивали вариант как потенциально влияющий на сплайсинг и сегрегационный анализ показал, что данный вариант находится в транс-положении с ранее описанной патогенной делецией. Все полученные данные расценивали данный вариант как «вариант неопределенного значения» согласно руководству по интерпретации данных, полученных методами массового параллельного секвенирования.
Для исследования влияния варианта на сплайсинг нами был проведен функциональный анализ с использованием системы экспрессии минигена в C19orf12 в модельной клеточной линии. В результате было показано, что вариант с.193+5G>A приводит к пропуску второго экзона мРНК C19orf12, что приводит к сдвигу рамки считывания и формирования укороченного белка с утратой его функции.
Результаты данной работы были опубликованы в журнале Neurogenetics
Функциональный анализ вариантов c.1932-3A>G и c.2613+1G>A в гене KIAA1109
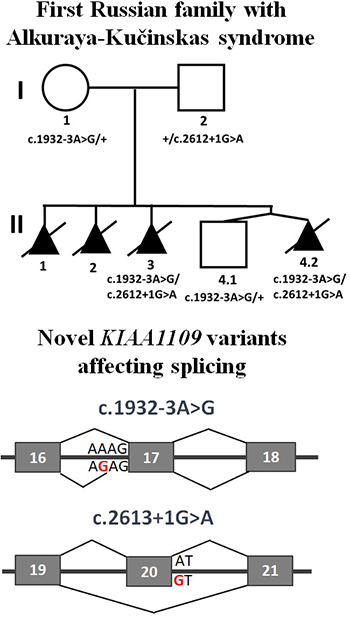
В лабораторию функциональной геномики обратилась врач-генетик МГНЦ Л.А. Бессонова с просьбой оценить патогенность вариантов нуклеотидной последовательности c.1932-3A>G и c.2613+1G>A в гене KIAA1109, обнаруженных у плода с множественными пороками развития. Пациентка с мужем обратились с третьей неудавшейся беременностью, прерванной на 14 неделе. У плода наблюдались вентрикуломегалия, артрогрипоз, отек мягких тканей головы и туловища и другие пороки развития. Кариотип родителей был в норме. Хромосомный микроматричный анализ (ХМА) ДНК плода не выявил отклонений. В дальнейшем в лаборатории «ГЕНОМЕД» было проведено полноэкзомное секвенирования ДНК плода, в результате чего были обнаружены два варианта с неизвестным клиническим значением в гене KIAA1109. Данный ген ранее не был ассоциирован с моногенными заболеваниями. Однако в литературе была описана гомозиготная мутация в данном гене у пациента с мальформацией Денди-Уокера, гидроцефалией, артрогрипозом, микрогнатией и другими нарушениями.
Проведенный нами биоинформатический анализ данного гена выявил его высокую консервативность, широкий спектр взаимодействий и высокую экспрессию в эмбриональном периоде. Кроме того, было известно, что модельные мыши с нокдауном данного гена не переживали эмбриональную фазу развития. В результате проведенного анализа был сделан вывод, что варианты в данном гене являются кандидатами для объяснения клинической картины пациентов. Проведенный нами сегрегационный анализ выявленных вариантов показал их транс-положение. Более того, ОТ-ПЦР анализ РНК выделенной из крови пациентов выявил, что замена c.1932-3A>G приводит к удлинению экзона 17 на 2 нуклеотида со сдвигом открытой рамки считывания и синтезом укороченного белка, а замена c.2613+1G>А приводит к пропуску 20 экзона, что приводит к пропуску 46 аминокислот в белке KIAA1109. Таким образом, оба исследуемых варианта нарушали нормальную функцию гена.
В процессе проведения нашей работы Gueneau et al. (2018) опубликовали работу, в которой описали новый синдром Алкурая-Кучинскас, вызываемый гомозиготными и компаунд-гетерозиготными мутациями в гене KIAA1109. Фенотипические проявления данного синдрома хорошо соответствовали выявленным у пациентов. Проведенный авторами работы функциональный анализ позволил нам переклассифицировать исследуемые варианты как патогенные. И на основании этого, супругам была успешна проведена пренатальная диагностика четвертой беременности, окончившаяся рождением здорового ребенка.
Результаты данной работы были опубликованы в журнале Clinical Genetics
Наша команда








Разработано в Лаборатории Функциональной геномики ФГБНУ «МГНЦ»
По возникающим вопросам и предложениям пишите на почту mskoblov@gmail.com